







Históricamente, la industria farmacéutica ha enfocado principalmente la validación de la limpieza como un ejercicio de aplicación de protocolos. Como tales, estas actividades de validación de limpieza se establecieron generalmente en función de las expectativas reglamentarias de las observaciones durante las inspecciones y no en planes maestros con base científica o evaluaciones de riesgos. A principios de la década de 1990, la FDA, así como otras agencias reguladoras, comenzaron a ver la limpieza como un proceso que requería validación. Por tanto, la validación de la limpieza se asoció estrechamente con la validación del proceso.
La validación de limpieza se basó en el enfoque tradicional de validación de otros procesos, que utiliza protocolos preaprobados, con criterios de aceptación predeterminados y las tres operaciones estándar. Este enfoque se adoptó sin considerar si eran necesarios tres ciclos de validación de limpieza, si eran suficientes o si los criterios de aceptación predeterminados eran apropiados para este caso. Al mismo tiempo, la industria estaba discutiendo sobre cómo establecer estos criterios de aceptación predeterminados requeridos.
Como parte de las GMP de la FDA para el proyecto siglo XXI, a partir de 2001 muchas nuevas iniciativas provinieron de las agencias reguladoras y de la propia industria farmacéutica. Tres eventos importantes anteriores a este momento también dieron forma a la dirección de la industria farmacéutica y de la validación de la limpieza: la decisión de Barr Labs en 1993, la Guía sobre la validación de la limpieza de 1993 de la FDA y las revisiones propuestas de 1996 a las GMP.

Algunas iniciativas provenientes de la industria farmacéutica incluían ‘Lean manufacturing’, Six Sigma y la excelencia operacional (OpEx), surgidas de las presiones para reducir costos o acelerar la producción y abastecer mejor al mercado con medicamentos. Estas presiones del mercado empujaron a la industria hacia procesos más efectivos y eficientes que centrarían esfuerzos y recursos donde aportasen el mayor valor. Las iniciativas de los reguladores se habían centrado en mejorar la calidad del producto, en el desarrollo y fabricación de procesos y en el uso de evaluaciones de riesgos. Si bien las iniciativas de la industria farmacéutica y los reguladores pueden parecer distintas entre sí, se pueden combinar de manera muy natural.
Todas estas iniciativas han atraído a la industria hacia enfoques basados ?en la ciencia, en la evaluación de riesgos, en las estadísticas y en la rentabilidad para garantizar la seguridad del paciente y la calidad del producto durante el desarrollo y la fabricación farmacéutica. Dado que la limpieza es uno de los procesos críticos en la fabricación, su rendimiento y validación también pueden beneficiarse de todas estas iniciativas.
La siguiente es una breve enumeración (14) de los principales eventos que han influido en la tendencia actual de la validación de la limpieza.
EEUU Vs. Barr Laboratories
A principios de la década de 1990, varias inspecciones a Barr Laboratories, Inc. por parte de la FDA dieron como resultado repetidas objeciones. Frustrados, Barr Laboratories entablaron una demanda contra la FDA. La FDA respondió solicitando al tribunal una orden judicial contra Barr Laboratories. El resultado de esta batalla legal fue la ahora famosa sentencia contra Barr Labs, en la que el juez Alfred M. Wolin encontró méritos en todas las afirmaciones de la FDA contra Barr Laboratories y acordó que las GMP requieren la inclusión de la validación del proceso y de la limpieza. Por extensión, la decisión respecto a Barr Labs se aplicó a toda la industria. Al mismo tiempo, el juez Wolin criticó las GMP por ser vagas y conniventes con las quejas de Barr Laboratories sobre las resoluciones aparentemente caprichosas e impredecibles de la FDA (15).
Artículo de Eli Lilly sobre los límites de limpieza
Al mismo tiempo que la demanda de Barr Laboratories, la mayoría de las empresas farmacéuticas estaban pugnando por establecer límites para la validación de la limpieza que fueran aceptables para la FDA. En 1992, la Asociación de Fabricación de Productos Farmacéuticos realizó una encuesta entre sus miembros y encontró que se estaban utilizando 44 enfoques diferentes. Se establecían muchos límites como una fracción de una dosis farmacéutica o como un nivel de concentración en un lote (por ejemplo, ppm). Posteriormente, en 1993, dos científicos de Eli Lilly and Co. (17) publicaron un artículo describiendo un enfoque que incluía:
- No aparecerá más de 0.001 dosis de cualquier producto en la dosis máxima diaria de otro producto.
- No aparecerán más de 10 ppm de cualquier producto en otro.
- Ninguna cantidad de residuo quedará visible en el equipo después de realizados los procedimientos de limpieza.
El enfoque de Eli Lilly combinó los criterios de dosis y concentración y fue adoptado por muchas empresas, principalmente porque la FDA lo mencionó en su guía de validación de limpieza (1), escrita en respuesta a las críticas del juez Wolin. Otras agencias reguladoras adoptaron rápidamente estos criterios en su propia guía. Sin embargo, con el tiempo, muchas empresas encontraron que estos criterios eran difíciles de cumplir y comenzaron a recurrir a límites menos estrictos, como 0,01 de una dosis o 100 ppm.
Revisiones propuestas de las GMP por la FDA en 1996
En respuesta a las críticas del juez Wolin y las quejas en curso de la industria, la FDA propuso cambios a las GMP en 1996 (3). Estos cambios especificaban actividades de validación muy claras tales como como pruebas de uniformidad de mezcla, y validación de métodos analíticos. Además, la FDA especificó que ahora se esperaba que además de la penicilina, cierta tipología de compuestos como agentes citotóxicos u otros antibióticos, deberían fabricarse en instalaciones dedicadas. En el preámbulo de los cambios propuestos, la FDA afirmaba:
"La agencia se ha abstenido de establecer una lista de medicamentos o productos farmacéuticos que presentan un riesgo inaceptable, porque tal lista se volvería obsoleta rápidamente... La FDA espera que los fabricantes identifiquen cualquier medicamento que produzcan que presente el riesgo de contaminación e implementar las medidas necesarias para eliminar ese riesgo. La FDA reconoce que, dependiendo del producto farmacéutico, puede ser aceptable una variedad de medidas para eliminar la contaminación cruzada; sin embargo, puede haber situaciones en las que otra cosa que instalaciones o equipos dedicados no será suficiente".
Si bien la industria comenzó a cumplir con estos requisitos de validación propuestos, las dificultades para cumplir con el requisito de potenciales instalaciones dedicadas comenzaron a aumentar. Como se mencionó anteriormente, muchas empresas ya tenían problemas para implementar los criterios de Eli Lilly (por ejemplo, límites muy estrictos para productos de bajo riesgo).
Guía ICH Q9
La emisión de la ICH Q9 aportó unos principios básicos de gestión de riesgos de calidad (QRM) y ejemplos de herramientas para este QRM que podrían aplicarse a procesos farmacéuticos. Aunque se utilizó con éxito en muchas otras industrias antes de ICH Q9, el análisis de riesgo no era común en la fabricación de productos farmacéuticos. ICH Q9 contiene dos principios fundamentales de la gestión de riesgos de calidad:
- La evaluación del riesgo para la calidad debe basarse en conocimientos científicos y, en última instancia, estar vinculada a la protección del paciente.
- El nivel de esfuerzo, formalización y documentación del proceso de gestión de riesgos de calidad debe ser acorde con el nivel de riesgo.
ICH Q9 también ofreció un marco para implementar un proceso de gestión de riesgos de calidad (figura 1).
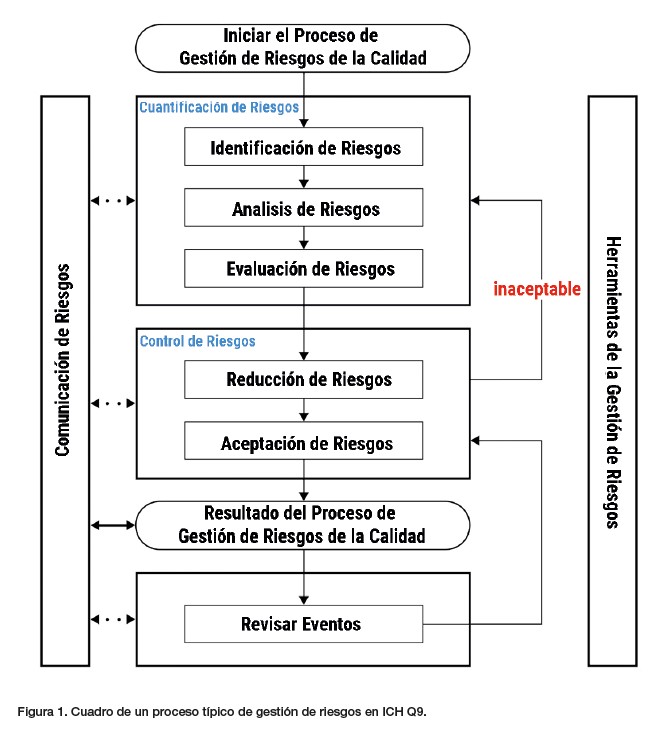
Al comparar la guía ICH Q9 con la revisión de 1996 de la FDA a las GMP, podemos ver una oportunidad de utilizar los dos principios básicos de actuación para abordar si sería necesario el requerimiento propuesto en 1996 sobre la dedicación de equipos y/o instalaciones para un producto determinado.
Fabricación de productos farmacéuticos basada en los riesgos (Risk-MaPP)
Al ir aumentando las preocupaciones por cumplir con los requisitos de dedicación, en 2004 se formó un equipo de toxicólogos farmacéuticos, higienistas industriales, profesionales de aseguramiento de la calidad, una pequeña empresa especializada en validaciones de limpieza y un representante de la FDA, para crear una guía para ser publicada por la Sociedad Internacional de Ingeniería Farmacéutica (ISPE). Varios de los autores de este artículo participaron en este esfuerzo. El propósito de esta guía era ayudar a las empresas a realizar una evaluación de riesgos de contaminación cruzada, caso por caso, para determinar los controles necesarios para garantizar la seguridad del paciente y la calidad del producto. Este enfoque caso a caso intentaba evitar la posibilidad de instalaciones dedicadas basadas simplemente en clases de productos (por ejemplo, todos los citotóxicos). Esta directriz se publicó en 2010 y se conoce como fabricación de productos farmacéuticos basada en los riesgos (Risk-MaPP). Para el enfoque Risk-MaPP es esencial un punto de partida apropiado para la evaluación de los procesos de limpieza.
Con el tiempo, se vio que el enfoque de Eli Lilly para establecer límites de validación de limpieza carecía de información toxicológica relevante, que resultaba en el establecimiento de límites más estrictos para algunos compuestos de bajo riesgo que para otros de alto riesgo (18). Estas discrepancias conllevaban una imagen confusa del riesgo real de contaminación cruzada e, incluso, podría resultar que requirieran dedicación compuestos de bajo riesgo (por ejemplo, aspirina en dosis bajas). Risk-MaPP introdujo el concepto de exposición diaria aceptable (ADE) como punto de partida para una evaluación de riesgos que podría evaluar los riesgos de contaminación cruzada de un producto y decidir si la dedicación es necesaria. Risk-MaPP definió el ADE como “una dosis que es poco probable que cause un efecto adverso si un individuo está expuesto, por cualquier vía, a esta dosis o por debajo de ella de por vida, todos los días".
El enfoque ADE es un criterio extremadamente estricto e inicialmente causó preocupación, considerando que muchas empresas ya habían tenido dificultades para lograr el enfoque de Eli Lilly, y este nuevo enfoque ADE podría ser aún más difícil de lograr. Sin embargo, un análisis comparativo del ADE con el criterio de dosis de 0,001 para 304 compuestos farmacológicos, determinó que el uso del criterio de dosis de 0,001 daba como resultado que los límites se establecieran demasiado bajos para el 85% de estos compuestos y en un grado significativo. Por otro lado, el enfoque de Eli Lilly no había sido lo suficientemente restrictivo para el 15% de estos compuestos (19). Por lo tanto, las dudas iniciales sobre el ADE eran infundadas y este enfoque, claramente, proporciona un alivio para muchas empresas con dificultades con el enfoque de Eli Lilly. Sobre todo, el enfoque basado en ADE permite una evaluación de riesgos que muestra los verdaderos márgenes de seguridad objetivos logrados en una situación de cambio de producto.
El ADE era un recambio obvio para el enfoque de Eli Lilly, al ser un parámetro basado en la salud derivado científicamente de todos los datos toxicológicos y clínicos disponibles para el compuesto y puede usarse para establecer límites apropiados para la limpieza y también para la exposición de los trabajadores. La figura 2 muestra una descripción general del proceso Risk-MaPP.

Documento conceptual de EMEA sobre instalaciones dedicadas para ciertos productos (6)
En su documento conceptual de 2005, la Agencia Europea de Medicamentos (EMEA) anunció que establecería qué otros medicamentos requerirían instalaciones especializadas, además de potentes sensibilizadores. El documento conceptual se centró principalmente en los denominados citotóxicos. La industria se opuso firmemente a esta propuesta, ya que entraba en conflicto con los principios de gestión de riesgos de calidad de ICH Q9 que se habían emitido el mismo año y también podría dar lugar a que muchas tipologías de compuestos tuvieran que fabricarse en instalaciones especializadas.
Proceso de validación FDA 2011: principios generales y prácticas.
En 2008, la FDA publicó un borrador de su guía de validación de procesos actualizada (10) para alinearse con el concepto del ciclo de vida del producto y con la guía existente de la FDA sobre ICH Q8-Q10. Esta nueva guía también describía conceptos directamente aplicables a la limpieza y la validación de la limpieza. Si bien esta nueva guía no aborda específicamente la limpieza, los elementos de la guía de validación del proceso se pueden enmarcar fácilmente como un enfoque de limpieza basado en la ciencia, el riesgo y las estadísticas. La FDA también ha declarado públicamente que la nueva guía se aplica a la validación de la limpieza (20).
Guía EMA sobre el uso de HBEL en instalaciones compartidas
En noviembre de 2014, la EMA (Agencia Europea del Medicamento) emitió una directriz que exige que las empresas revisen y evalúen los datos farmacológicos y toxicológicos de las sustancias activas individuales para determinar sus límites de exposición basados en la salud (HBEL), utilizando exposiciones diarias permitidas (PDE) para su uso como una herramienta de identificación de riesgos y para justificar los límites acumulados utilizados en la validación de limpieza (10). La EMA utiliza el término PDE para describir un HBEL y establece explícitamente que sus PDE se consideran equivalentes a los ADE. En esta guía, la EMA requería que todas las empresas tuvieran los HBEL adecuados a partir de diciembre de 2015.
El requerimiento de utilizar HBEL también se incorporó en el anexo 15 en octubre de 2015 y se eliminaron las referencias anteriores al uso de una dosis de 0,001 y 10 ppm para la validación de la limpieza (21). El Plan de cooperación para la inspección farmacéutica (PIC / S), que ahora incluye a 54 organismos de todo el mundo incluida la FDA, integraron el Anexo 15 de la EMA en su propia serie de documentos (22). En 2018, el PIC/S también adoptó literalmente las directrices GMP que la EMA había modificado en 2014, cuando se introdujo el concepto de valores límite basados en la ciencia. Entonces, en efecto, el uso continuo de la dosis de 0.001 y 10 ppm para los límites de aceptación de validación de limpieza ya no se reconoce como apropiado o aceptable en la UE, los EE. UU y la mayor parte del mundo (15).
Estándares ASTM y su significado
ASTM International es una organización global que ofrece un desarrollo totalmente transparente de estándares voluntarios de consenso que cuenta con más de 30.000 miembros de más de 140 países. Los miembros de ASTM participan en equipos para desarrollar guías, prácticas, métodos y especificaciones estándar utilizando su experiencia global, ciencia e ingeniería para mejorar el desempeño en manufactura y materiales, productos y procesos, sistemas y servicios. Los miembros de ASTM incluyen empresas, gobiernos e individuos interesados ??que colaboran de forma abierta y transparente en comités técnicos para desarrollar normas. Todos los estándares son votados por los miembros del comité de gobierno y todos los votos negativos deben resolverse satisfactoriamente con el votante hasta que se alcance el consenso. Actualmente, ASTM tiene más de 12.500 estándares que se utilizan en todo el mundo. ASTM formó el Comité E55 sobre la fabricación de productos farmacéuticos en 2003 y, desde entonces, se han publicado o se están desarrollando varias normas relacionadas con la validación de la limpieza. Como una de las pocas organizaciones de desarrollo de estándares de consenso, ASTM satisface los requisitos de la Ley Nacional de Transferencia y Avance de Tecnología de los EEUU de 1995, por lo que la FDA puede reconocer y adoptar los estándares de ASTM.
ASTM E3106 (12)
Esta nueva norma aplica el enfoque del ciclo de vida a la validación del proceso de limpieza, que incluye el desarrollo, la calificación y la verificación de los procesos de limpieza. Es aplicable a productos farmacéuticos (incluidos ingredientes farmacéuticos activos -API-, formatos de dosificación, medicamentos de venta libre, productos veterinarios, biológicos y suministros clínicos) y también es aplicable a otros productos de salud, cosméticos y de consumo. Este estándar utiliza el HBEL (ADE) para evaluar el riesgo para los pacientes de la limpieza de equipos de fabricación y dispositivos médicos y se alineará con el estándar E3219.
La nueva norma ASTM E3106 incide mucho más en la aplicación de la ciencia y el riesgo en las etapas de identificación y análisis de riesgos, incluido el desarrollo del proceso de limpieza, selección de métodos analíticos, análisis de riesgos de los datos históricos, análisis de riesgos de los SOP, etc. (figura 3). El trabajo apropiado en estas primeras etapas puede representar reducciones en el nivel de esfuerzo, formalización y documentación del proceso de validación general, permitiendo la selección de métodos analíticos basados en riesgos (por ejemplo, inspección visual) y simplificando la introducción de nuevos productos.

ASTM E3219 (13)
El propósito del E3219 es estandarizar la derivación del HBEL. Tanto Risk-MaPP como la Guía sobre límites de exposición basados en la salud de la EMA contienen una guía limitada y sus cálculos son diferentes en algunos aspectos. El E3219 describe los procedimientos científicos para evaluar e interpretar los datos toxicológicos y clínicos de un API y cómo utilizar los datos para derivar un HBEL que puede usarse para la evaluación de la contaminación cruzada durante la fabricación de diferentes productos en las mismas instalaciones de fabricación. El E3219 debe usarse para calcular y documentar un HBEL para API (incluidos los biológicos), productos intermedios, agentes de limpieza, excipientes u otros productos químicos que se hayan identificado como riegos potenciales en la limpieza. El E3219 hace referencia y está destinado a ser utilizado junto con la Guía Estándar ASTM E3106.

El E3219 también establece las cualificaciones que se requieren de las personas que establecen el HBEL (experto cualificado) y los requisitos mínimos de documentación para la derivación. El estándar también está destinado a ser utilizado por inspectores reguladores (por ejemplo, FDA, EMA, etc.) al evaluar los HBEL que encuentren durante las inspecciones.
Estándares ASTM en proceso de desarrollo
Actualmente existen varias normas ASTM adicionales en diversas etapas de desarrollo sobre temas importantes en la validación de la limpieza. Dos de estos son el elemento de trabajo ASTM 64938 y el elemento de trabajo ASTM 67425.
Resumen
Las nuevas normas ASTM discutidas anteriormente se pueden utilizar para crear enfoques basados en la ciencia y los riesgos para la limpieza y su validación. Estos estándares ofrecen una guía para realizar cambios basados en datos en los enfoques de validación de limpieza que pueden minimizar la complejidad, reducir los costes y acortar los procesos, al tiempo que brindan una mayor probabilidad de que la limpieza de los equipos de fabricación farmacéutica sea efectiva. Al implementar un enfoque verdaderamente basado en la ciencia, como con el uso de los HBEL para el análisis de riesgos, con las evaluaciones de riesgos adecuadas y con el desarrollo del proceso de limpieza, se puede desarrollar fácilmente un programa de limpieza simplificado que garantice, al mismo tiempo, la seguridad del paciente y la calidad del producto, aligerando la carga regulatoria sobre la industria. Los próximos artículos de esta serie proporcionarán un análisis más detallado de E3106 y E3219.
Referencias:
| Nombre | Andrew Walsh, Thomas Altmann, Joel Bercu, Ph.D., Alfredo Canhoto, Ph.D., David G. Dolan Ph.D., Andreas Flueckiger, M.D., Igor Gorsky, Jessica Graham, Ph.D., Ester Lovsin Barle, Ph.D., Ovais Mohammad, Mariann Neverovitch, y Osamu Shirokizawa |
|---|---|
| Empresa | Halltech |
| Cargo |

























Política de privacidad | Cookies | Aviso legal | Información adicional| miembros de CEDRO



















