





Alonso Fernández Nistal
BU Head de Gastrointestinal y Vacunas, Takeda

Pere Mañé
CEO, Suanfarma

Almudena del Castillo
Directora General en España, QbD Group













Guía rápida para el entendimiento clínico

Mis muy estimados lectores. Desde hace meses, me he negado a escribir sobre la COVID. Bastantes doctos y sabios tenemos sobre el tema y me seguiré, por ahora, negando a ello. Eso sí, en este artículo daremos una guía rápida para entender lo que es la clínica de medicamentos, tan de moda y como consecuencia de la muy deseada y esperada vacuna anti COVID.
Revisaremos en el presente artículo los conceptos y fases fundamentales para, evidentemente, no hacernos expertos (ya tenemos bastantes), sino para entender un poco mejor lo que nos van a inyectar, esperemos, que en breve espacio de tiempo.
Seguridad y Eficacia
Axioma, esta vez a demostrar, e hipótesis de partida y base fundamental de la clínica, es que el fármaco reúne dos cualidades básicas:
- Seguridad
- Eficacia
Es decir, dicho en román paladino, que cura y no mata.
Los estudios clínicos deben estar diseñados para responder a unas preguntas esenciales, según la FDA norteamericana:
- ¿Funciona el medicamento?
- ¿Es mejor que los tratamientos que ya se usan actualmente?
- Si no es mejor, ¿es igual de eficaz y causa menos efectos secundarios?
- ¿Funciona en algunas personas que no responden a los tratamientos actuales?
- ¿Es seguro el nuevo tratamiento? No existe tratamiento o procedimiento, incluso ninguno de los que se usan normalmente, que no conlleve un riesgo. Sin embargo, ¿son mayores los beneficios el nuevo tratamiento que los riesgos?
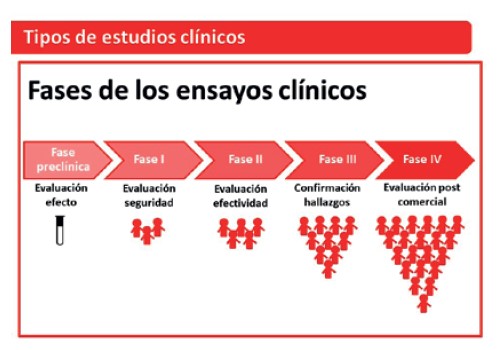
Para responder a estas cuestiones, así como para determinar la dosis y pautas de administración adecuadas, se diseñan y ejecutan las diferentes fases por las que discurre un ensayo clínico oficial para un nuevo fármaco (ver figura 1):
- Estudios preclínicos o de laboratorio.
- Estudios clínicos en fase I: determinación de la dosis tolerable. Seguridad
- Estudios clínicos en fase II: determinación de la dosis terapéutica.
- Estudios clínicos en fase III: confirmación de la dosis terapéutica.
- Estudios clínicos en fase IV: confirmación de la eficacia y seguridad en la población general.
La esencia de cada fase, lo que queremos demostrar a las agencias reguladoras para que nos aprueben el fármaco/vacuna, viene muy claramente descrita en la figura 2.
Antes de seguir adelante con el artículo debemos reflexionar sobre tres puntos muy importantes a tener en cuenta y llevar en nuestro mesencéfalo:
- Los ensayos clínicos se realizan sobre individuos, personas…
- Se realizan sobre personas sanas y enfermas.
- No hay ningún fármaco que no tenga efectos secundarios.
- No hay ensayo clínico sin riesgo, no existe el riesgo cero.
- Actualmente el coste medio de un ensayo clínico se sitúa, en torno a 1.800 millones de euros.
- Un ensayo debe ser llevado a cabo, siempre que los beneficios previstos justifiquen los riesgos.
Fase cero: estudios preclínicos o de laboratorio
Es muy importante remarcar que los estudios clínicos se realizan únicamente después de que los hallazgos en los estudios preclínicos hayan indicado que el tratamiento o medicamento nuevo podría ser seguro y que funcionará en las personas. El objetivo de esta fase inicial son dos:
- Determinar la utilidad.
- Valorar la toxicidad del fármaco.
Para demostrar esto, los estudios preclínicos o estudios de laboratorio suelen incluir:
- Estudios sobre cultivos celulares: son test que se realizan sobre células in vitro.
- Estudios en animales: los tratamientos que pasan los test celulares pasan a ser testados en animales vivos, con el fin de proporcionar una idea de la dosis máxima permitida y la dosis letal 50 (aquella que produce la muerte en el 50% de la población).
Entiendo que nos cause un impacto el uso de animales, yo lo he visto en primera línea, pero debemos pensar que, mientras no haya modelo in vitro/ordenador que puedan sustituir con garantías esta fase, debemos seguir adelante con ello. Pensemos los cientos de miles de muertos que llevamos sobre nuestros hombros durante esta pandemia…
Los estudios preclínicos proporcionan bastante información útil, pero no todo lo que es necesario saber. Los humanos y los animales son diferentes en la forma en cómo absorben, procesan y eliminan los medicamentos. Un tratamiento que funciona en un animal puede que no funcione en las personas. Asimismo, puede que en las personas surjan efectos secundarios y otros problemas que no se presentaron cuando el tratamiento se usó en animales, si bien las curvas de interrelación tienen bastante correlación humano – animal. Una vez terminada la fase cero de forma exitosa, la empresa investigadora debe pedir una autorización a las agencias regulatorias, para seguir adelante con las siguientes fases, el famoso IMPD (Investigational Medicinal Product Dossier).
Según la European Investigational Medicinal Product Dossiers, el Dossier de Medicamentos en Investigación (IMPD) es documento que reúne varios datos relacionados con los medicamentos en investigación (IMP) requeridos, siempre que la realización de un ensayo clínico se lleve a cabo en uno o más estados miembros de la Unión Europea.
El IMPD incluye resúmenes de información relacionada con la calidad, fabricación y control de cualquier IMP (incluido el producto de referencia y el placebo) y datos de estudios clínicos y no clínicos.
Se puede encontrar orientación sobre los expedientes de IMP en la comunicación de la Comisión Europea titulada ‘Orientación detallada sobre la solicitud a las autoridades competentes de autorización de un ensayo clínico sobre un medicamento de uso humano, la notificación de modificaciones sustanciales y la declaración de finalización del juicio’.
La guía se basa en el Reglamento (UE) nº 536/2014 sobre ensayos clínicos de medicamentos de uso humano (deroga la Directiva 2001/20 / CE) sobre la aproximación de las leyes, reglamentos y disposiciones administrativas de los estados miembros, relativas a la implementación de buenas prácticas clínicas en la realización de ensayos clínicos sobre medicamentos de uso humano (también denominada comúnmente ‘Directiva sobre ensayos clínicos’) (figura 3).

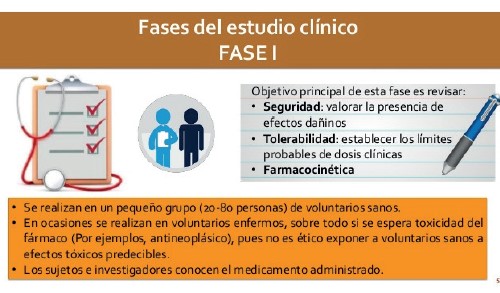
Fase I: determinación de la dosis tolerable. Seguridad
La fase I (figura 4) es en la que se involucra a personas por primera vez, generalmente enfermos en un grupo reducido de unos 20 a 80 individuos. Su objetivo esencial es la determinación de la máxima dosis permitida por el organismo, sin causar efectos secundarios graves y ver el comportamiento del fármaco en el organismo (farmacocinética).
En la fase I, el primer grupo pequeño recibe una dosis baja del tratamiento y es observada minuciosamente, de tal manera que si solo aparecen efectos secundarios menores, el próximo grupo pequeño de participantes recibe una dosis más alta. Y así el proceso continúa hasta que se encuentra la dosis más adecuada y mientras se mantenga un nivel aceptable de efectos secundarios.
Fase II: determinación de la dosis terapéutica
Si en la fase I se determina que un tratamiento nuevo es seguro, entonces se procede con la fase II (figura 5) para determinar su eficacia frente a la patología, cuyas características fundamentales son las siguientes:
- Entre 25 y 100 personas con la misma patología reciben el nuevo tratamiento.
- Se utiliza la dosis y el método que se determinaron ser los más seguros y efectivos en la fase I del estudio.

Fase III: confirmación de la dosis terapéutica
El objetivo principal de la fase III es confirmar la dosis terapéutica final más adecuada, y que presente los menores efectos adversos serios posibles, si bien se sigue evaluando la seguridad del fármaco. Hay que tener en cuenta que las fases de los estudios clínicos son dinámicos e interrelacionados íntimamente entre ellos.
Normalmente se introducen en el estudio más de 20.000 pacientes, que se acogen a un programa y que se escogen al azar. Tanto el investigador como el paciente desconocen cuál es el tratamiento que está recibiendo (ciego) y parte de los pacientes son inoculados con placebo (doble ciego aleatorio).
Llegados a este punto, debemos remarcar que los principales players en los estudios clínicos son:
- Promotor del ensayo.
- Investigador principal, que es el responsable en el centro. Es un clínico experto.
- Monitor, que verifica que todo el proceso se realiza según las normativas y protocolos aplicables.
- Pacientes.
- Comité ético, acreditado por la autoridad competente, quien verifica las correctas prácticas de ensayos clínicos y aprueban el IMPD.
- Centros de ensayo, generalmente se usan varios en paralelo (multicéntricos).
- Agencia regulatoria.

Fase IV: confirmación de la eficacia y seguridad en la población general
Los medicamentos aprobados se mantienen bajo observación durante un largo tiempo mediante los estudios en fase IV (figura 6), ya que pueden aparecer al cabo del tiempo efectos adversos nuevos o exacerbarse algunos ya detectados durante las fases clínicas. Se hace el seguimiento de más de 100.000 pacientes.
Una vez que hemos repasado las fase, ¿cuánto tiempo se necesita para llevar a cabo todas las fases?
Generalmente se necesitan unos 10-12 años para ejecutar desde la fase I hasta la III incluida, tal como podemos ver segmentado en la figura 7.
En el esquema de la figura 8 podemos ver el marco legal aplicable en nuestro país y su evolución desde el año 2001 hasta la actualidad.
Pues sí lectores, ahora os preguntaréis cómo en 9 meses tenemos la vacuna de la COVID, si generalmente se tarda más de 10 años… Las respuestas que las den Pfizer, Moderna, AstraZeneca, etc. De lo que sí estoy totalmente convencido es de que son compañías muy serias y que serán seguras y eficaces. Yo me vacunaré.
Y si necesitáis algo más, ya sabéis dónde estoy.



| Nombre | Eduardo Sanz |
|---|---|
| Empresa | Pharmaceutical Industry |
| Cargo | Senior Advisor |

























Si continúas navegando, aceptas su uso.
Más información





Política de privacidad | Cookies | Aviso legal | Información adicional| miembros de CEDRO